Valuing Biotech & Pharma Companies
The valuation of privately held biotechnology and pharmaceutical companies is generally very challenging because of the long development cycles and uncertainties of clinical testing and product registration outcomes — even before market acceptance and competition is considered. Such companies are typically in the early stages of the drug development process, meaning that products are in Preclinical Development, Phase I or Phase II stages. Most such companies do not have significant revenues or positive earnings, and their assets will generally consist of cash or short-term investments and intellectual property that is difficult to assess, making it difficult or impossible to apply most traditional valuation methods.
The use of a classic Discounted Cash Flow analysis for a biotechnology company with products in an early stage of development is not meaningful since projected future revenues will be contingent on the products successfully going through clinical trials and product registration — where there is uncertainty and thus always less than 100% probability of success. Many analysts try to adjust for this by adding a high risk premium to the cost of capital used, but we believe that this approach is misleading.
For a more detailed discussion about the drug development process, please see our primer in the Resources section.
Risk-Adjusted Net Present Value (rNPV)
Over the past two decades, Risk-Adjusted Net Present Value or rNPV analysis has emerged as a better tool. With rNPV analysis the expected future cash flows are calculated by first estimating the future cash flows without risk and then applying a probability to that cash flow. For example, if overall average pharmaceutical development probabilities (Phase I = 52%, Phase II = 29%, Phase III = 58%, FDA Approval = 90%) are applied, the compound probability from Phase I to Product Approval will be 7.9%. Therefore any future net cash flows from when a product is in the market would be multiplied by this percentage to arrive at an expected net cash flow. These cash flows would then be discounted to present value using typically an industry-based cost of capital representative of "systematic risk" rather than an estimated cost of capital for the company being valued, i.e., no company specific risk premium is included.
However, the rNPV method has also come under criticism for providing value estimates that are unrealistically precise and focused on average numbers. In the real world there is never a 90% product approval — it is either approved or not.
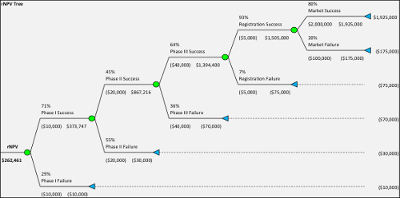
You can try out this methodology yourself with our interactive rNPV Calculator.
Monte Carlo Simulation
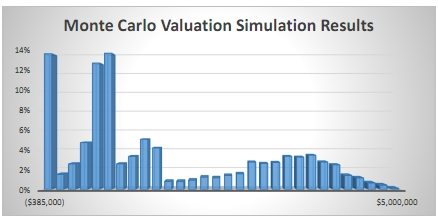
Monte Carlo Simulation has come to be more and more widely used, enabled by more powerful computers and better software, to analyze these problems. With a Monte Carlo simulation, thousands or millions of potential outcomes can be tested for the development and market introduction of a pharmaceutical product, providing a distribution of outcomes which can be further analyzed — and often reveals a different picture from rNPV alone.
An average value provided by an rNPV analysis can be misleading because outcomes are more likely to be either a zero value or a high value, with the average value unlikely to occur in reality. For an introduction to Monte Carlo simulations please visit www.probabilitymanagement.org.
Real Options Analysis
To further incorporate the dynamic effects of a drug development process we can also utilize a Real Options analysis to complement an rNPV or Monte Carlo analysis. The Real Options analysis recognizes the inherent optionality in such a process — for example, that a decision to undertake costs for a Phase II trial does not need to be made until the results from the Phase I trial are known. Projects that have a negative net present value in an rNPV analysis can therefore have a positive net present value when analyzed with a Real Options methodology. A Real Options analysis is often performed using a binomial lattice options technique, although Monte Carlo simulation can also be used.
Real Options analysis is a valuable tool for evaluating biotechnology and pharmaceutical projects because it allows decision-makers to account for the uncertainty, flexibility, and staged investment nature inherent in drug development. Key aspects of Real Options analysis include the following:
Flexibility and Decision-Making Under Uncertainty - Research & development for drugs is inherently risky, with high uncertainty about the outcomes at each stage of development. Real Options analysis enables companies to value the flexibility they have in making decisions as the project evolves, such as whether to continue, delay, or abandon a project based on new information. This is analogous to financial options where the holder has the right, but not the obligation, to take certain actions.
Staged Investment Approach - Drug development typically involves multiple stages (e.g., preclinical, Phase 1, Phase 2, Phase 3, and regulatory approval), each requiring significant investment. Real Options analysis values the project in stages, considering that a company can decide at each phase whether to invest further or not, depending on the results of the previous stage. This approach reduces the downside risk while allowing companies to capitalize on upside potential if the project progresses successfully.
Valuing Future Opportunities - Real Options analysis helps in valuing future opportunities that a pharmaceutical project may create, such as the potential to expand into new markets, develop additional drug indications, or combine with other therapies. These potential opportunities can be valued as options that add to the overall project value.
Decision Trees and Option Valuation - In practice, Real Options analysis often involves constructing decision trees that map out possible future paths and outcomes of a project. Each branch of the tree represents different decisions (e.g., continuing development, pausing, or terminating the project) and their associated probabilities. The value of these options is then calculated using techniques similar to those used for financial options, such as the Black-Scholes model or binomial models.
Mitigating Risk - By incorporating Real Options analysis, pharmaceutical companies can better understand and mitigate the risks associated with R&D projects. For example, the option to delay investment until more information is available (such as the results of early-phase trials) can be highly valuable in managing uncertainty and making more informed investment decisions.
A traditional Net Present Value (NPV) analysis often underestimates the value of pharmaceutical projects because it does not account for the flexibility to make decisions at various stages. Real Options analysis, on the other hand, provides a more dynamic and realistic valuation by incorporating the value of this flexibility.
Examples of Valuation Engagements
| Therapeutic Segment | Development Stage | Valuation Purpose |
|---|---|---|
| Oncology | IND | 409A |
| Metabolical | Phase I | Transaction |
| Respiratory | Preclinical | 409A |
| Cystic Fibrosis | Phase I | Gift Tax |
| Wound Care | Phase III | Transaction |
| Neurology | IND | 409A |
| Generics | Marketed | Transaction |
| Oncology | Phase II | Estate |
| Neurology | Phase II | Purchase Price Allocation |
| Radiotherapeutics | Phase II | Transaction |
| Infectious Diseases / Oncology | Phase II | Transaction |
| Oncology | Preclinical | Transaction |
| Various 505(j) and 505(b)(2) | IND | Transaction |
| Vaccines | IND | 409A |
| Autoimmune Disorders | Phase I & II | 409A |
| Respiratory | Phase I | 409A |
| Generics | Marketed (503B) | Transaction |
| Immunotherapy / Oncology | IND | Transaction |