Corporate Finance
Business Valuations
Mergers & Acqusitions
United States • Europe • Asia
Biotechnology & Pharma Company Valuations
The valuation of privately held biotechnology and pharmaceutical companies is generally very challenging because of the long development cycles and uncertainties of clinical testing and product registration outcomes even before market acceptance and competition is considered. Such companies are typically in the early stages of the drug development process, meaning that products are in Preclinical Development, Phase I or Phase II stages. Most such companies do not have significant revenues or positive earnings, and their assets will generally consist of cash or short-term investments and intellectual property that is difficult to assess, meaning that it is difficult or impossible to apply most traditional valuation methods.
The use of a classic Discounted Cash Flow analysis for a biotechnology company with products in an early stage of development is not meaningful since projected future revenues will be contingent on the products successfully going through clinical trials and product registration where there is uncertainty and thus always less than 100% probability of success. Many analysts try to adjust for this by adding a high risk premium to the cost of capital used, but this approach is misleading.
Instead over the past two decades Risk-Adjusted Net Present Value or rNPV analysis emerged as a better tool. With rNPV analysis the expected future cash flows are calculated, by first estimating the future cash flows without risk and then applying a probability to that cash flow. For example if typical pharmaceutical development probabilities (Phase I = 71%, Phase II = 45%, Phase III = 61%, Product Registration = 93%) are applied the compound probability from Phase I to Product Approval will be 18%. Therefore any future net cash flows from when a product is in the market would be multiplied by this percentage to arrive at an expected net cash flow. These cash flows would then be discounted to present value using typically an industry based cost of capital representative of “systematic risk” rather than an estimated cost of capital for the company being valued.
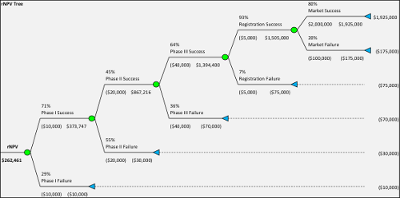
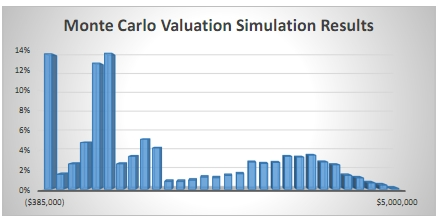
However the rNPV method has also come under criticism for providing value estimates that are unrealistically precise and concrete as well as focused on average numbers. In the real world there is never a 90% product approval – it is either approved or not. Instead Monte Carlo Simulation has come to be more and more used with more powerful computers and better software to analyze these problems. With a Monte Carlo simulation thousands or millions of potential outcomes can be tested for the development and market introduction of a pharmaceutical product, providing a distribution of outcomes which can be further analyzed and often reveal a different picture. An average value provided by an rNPV analysis can be misleading because outcomes are more likely to be either a zero value or a high value, with the average value unlikely to occur in reality. For an introduction to Monte Carlo simulations visit www.probabilitymanagement.org.
To further incorporate the dynamic effects of a drug development process we can also utilize a Real Options analysis to extend an rNPV or Monte Carlo analysis. The Real Options analysis recognizes the inherent optionality in such a process, for example that a decision to undertake costs for a Phase II trial does not need to be made until the results from the Phase I trial are known. Projects that have a negative net present value in an rNPV analysis can therefore have a positive net present value when analyzed with a Real Options methodology. A Real Options analysis is often performed using a binomial lattice options technique although Monte Carlo simulation can also be used.
409A & ASC 718 Valuations
Windeye Partners performs business valuations for biotechnology, pharmaceutical and other life science companies that plan to provide option or stock incentives to employees and need to comply with the U.S. tax code (i.e. Section 409A) and related AICPA rules. For an overview please see our 409A Valuations page. In addition, we assist clients in determining the expense of options and similar equity instruments issued to employees for financial reporting purposes (ASC 718).
Intellectual Property Valuations
Windeye Partners performs intellectual property valuations for biotechnology, pharmaceutical and other life science companies with respect to patented compounds and products in connection with corporate spinoffs or transfers to other entities. In such situations a valuation may be required because of IRS regulations or needed because of resulting ownership changes. For an overview please see our Intellectual Property Valuations page.
Investment Fund Valuations
Windeye Partners assists investment funds and family offices with valuation analysis and complex financial models in connection with investments in biotechnology, pharmaceutical and other life science companies, as well as in connection with financial reporting of the fair value of privately held or thinly traded securities (ASC 820).
Examples of Valuation Engagements
Transaction
Transaction
Transaction
409A
409A
409A
Transaction
Transaction
Infectious Diseases/Oncology
Oncology
Various 505(j) and 505(b)(2)
Vaccines
Autoimmune Disorders
Respiratory
Generics
Immunotherapy/Oncology
Phase II
Preclinical
IND
IND
Phase I and II
Phase I
Marketed (503B)
IND
Biotechnology & Pharma Valuation Primer
The Drug Development Process
The drug development process in the U.S. is a lengthy and cost-consuming process, primarily because there is a focus on making sure that all drugs that gain approval for sale and marketing are not only safe for use but also effective for the intended (and claimed) purpose. Prior to the creation of the modern U.S. Food and Drug Administration (FDA) in 1938 just a small handful of drugs received approval for sale and marketing in the United States. Through the 1930’s and 1940’s only about four compounds per year were approved. This number grew to an average of 10 annually through the 1990’s and peaked at 53 in 1995. This increase is commendable, but one should keep in mind that still through 2013 less than 1,500 new drugs in total had been approved for sale in the United States. After a period of declining and lower levels of drug approvals, recent years have seen an increase with a new record of 59 drugs approved in 2018.
How A Drug Is Developed
To begin, almost any drug development process must proceed through several stages in order to create, refine, manufacture, market and sell a product that is safe, efficacious and has passed all regulatory approval requirements in the United States. The exact number of stages depends on categorization and classification of the different activities involved and different schools of thought put it between 6 and 10 separate stages but what is more important is the aim and accomplishments of the functional stages of completion rather than the exact number or grouping, which we will summarize below. As a reminder, before we begin, drug development is not a fast or an inexpensive venture consuming a great deal of time and resources which may partly indicate why many pharmaceuticals may carry such high price tags.
Stage 1 – Identification and Validation
This is the time at which small molecule or biological compounds, often a gene or protein, are identified and selected for development as leads for any given disease. Following the identification of such molecules the researchers will then also confirm that the molecule(s) are indeed involved with/related to the disease in question. If the molecule shows promise as a therapeutic it must then be characterized as to size, strength(s) and weakness(es), as well as undergoing thorough throughput screening regarding conditions to maintain function including acute, repeated dose, genetic and reproductive toxicity, carcinogenicity, and bioactivity studies. The formulation must remain potent, sterile and safe (nontoxic) to continue. The improvement or refinement of hit compounds that have been identified as “leads” are then focused on in terms of efficacy, exposure safety, potency and stability. At this stage it is not uncommon for developers to have several compounds on hand.
Stage 2 – Preclinical Development
At this phase the testing of the “lead” candidates is conducted which is primarily broken down into two stages: in vitro and in vivo testing; in vitro refers to the interaction of the molecules in a test tube and lab setting while in vivo refers to testing on animals and other living cell cultures. Such evaluations have also come to be labelled as “IND enabling studies” since the end result and goal of the testing in the preclinical stage is completing the data required and preparation of an Investigational New Drug (IND) application.
While efficacy is beginning to be established safety is a primary concern as preclinical studies will be prohibited by the FDA to move into clinical trials without extensive data on safety. In addition to all molecule composition and testing to date, the IND must include a full scale-up manufacturing of the “leads” for human clinical trials for application and submission to the FDA. At this time researchers will be dealing with anywhere from 1 to 5 candidates and not the hundreds with which it may have begun.
Stage 3 – Investigational New Drug Application (IND)
The third step involves submitting a request to the FDA for authorization to administer such experimental drug or biological product in conducting clinical trials on humans. The FDA will scrutinize the results of preclinical testing focusing on safety, side effects, chemical structure and relationship to promising results as well as possible manufacturing processes for the drug. The application and authorization must precede the start of clinical trials and only if the FDA approves the IND can a company proceed. At this stage actual clinical trials are commenced, and it is important to note that once an IND is filed and approval issued this is the point at which a patented drug's 20-year exclusivity period begins.
Stage 4 – Clinical Trials
Phase I Clinicial Trial
Often referred to as the “Human Pharmacology” stage, this is the first-time that studies are conducted to determine the safety and pharmacokinetics (which is concerned with the movement of drugs in the body) of the drugs in what is generally a limited number of healthy humans. The primary focus will be on safety regarding how the drug is absorbed and eliminated from the body, possible side effects of increasing, continued or maximized doses and whether it is producing the desired results.
Phase II Clinical Trial
Also known as the “Therapeutic Exploratory” stage as it encompasses studies conducted to evaluate the effectiveness of a drug for a particular disease or signs or symptoms in patients with the condition under study. In addition, the largest difference between Phase I and Phase II trials is that the patient population is increased from 15-30 to several dozen or even 100+ and secondly, the patient pool is not healthy humans but those afflicted with the ailment of focus. As earlier, safety remains a preeminent concern however the focus begins to shift towards efficacy. During Phase II trials optimal dosing is also examined. At this point we should take a look back and recognize that most NME’s fall off the testing radar during Phases I and II, leaving only a small remaining number of compounds viable for Phase III.
Phase III Clinical Trial
In what is known as the “Therapeutic Confirmatory” stage of the Clinical trials, safety continues to be a priority, but the primary intent is to gather additional information to evaluate the overall risk-benefit trade-off posed by the use of the drug in afflicted patients. In other words, to what degree is the compound able to alleviate or at best eradicate the symptoms or cause(s) of the disease that the use of it is a positive to those suffering? Phase III trials are designed by the researchers but must be approved by the FDA prior to commencement and generally contain formal guidelines and defined endpoints or milestones to determine the success or failure of a proposed drug compound. Phase III trials include a much larger patient base generally in the hundreds or in some cases, even thousands and are generally the longest in duration and most costly of all clinical trials. At this point researchers are not only focused on determining definitive efficacy but also future manufacturing potential if the results are encouraging. If the experimental drug meets its designated endpoint(s) and can prove to be safe for use by patients the researchers begin the arduous task of filing for its approval.
Stage 5 – New Drug Application (NDA) Filing
The New Drug Application (or in the case of a biologic product a Biologic License Application – BLA) is submitted to the FDA for approval of use and must include all research and safety data observed during the prior steps. The application also includes manufacturing scale-ups and marketing in designated countries. If the NDA is accepted, a PDUFA (Prescription Drug User Fee Act) date is established 10 months from that date at which time the FDA is expected to make a decision.
Stage 6 – PDUFA Date and Decision
The FDA and its related agencies generally wait until the PDUFA date to release their findings and decision. There are three potential outcomes: outright denial, request additional information through issuance of a Complete Response Letter (CRL) or approval. Outright denial is rare. Receipt of a CRL basically states what was lacking in the findings and or application that prevented the drug from being approved and offers suggestions on how to remedy the situation. This often requires the researchers to conduct additional studies and or alter the proposed manufacturing process in order to gain FDA approval. The NDA is then resubmitted and If the drug is then approved by the FDA it becomes immediately available for commercial production and marketing.
Stage 7 – Phase IV Clinical Studies/Post-Marketing Trials
Even after a drug is approved and sold in the U.S. market the drug developer cannot simply focus on the manufacture and marketing of the compound and it is not uncommon for the FDA to request long-term safety studies. Studies are generally designed to provide additional information on the drug which may include the risks, benefits, use and optimal dosing. The studies may be voluntarily conducted or as required by the FDA and other regulatory agencies.
Summary
As we can see developing a drug is not a simple, quick or affordable venture process. A typical estimate from the point of identifying compounds to drug delivery to the public is 12 years, but this can vary depending on the complexity of the compound. According to a recent study by Tufts University Center for the Study of Drug Development the average cost associated with the process is $2.6 billion. Only 5 in 5,000 drugs (0.1%) that enter preclinical testing progress to human testing, and only one of these 5 drugs that are tested in humans is approved. Hence, the odds of a new drug making it to market are approximately 1 in 5,000 (0.02%). For this and other reasons there has been an increasing emphasis on the idea of private or public companies pairing with and working in conjunction with universities, governments, and other entities in the pharmaceutical and other (genetic, technology, NGO research and think-tanks) fields to collaborate resources and share costs to make it a more affordable and efficient process yielding more effective prescription drugs at a faster pace and a cost that is accessible to a greater number of consumers.
The Drug Development Timeline
The timelines for initial drug identification and validation as well as preclinical development have significant variations, some of which may result from a lack of funding or priorities, or collaboration with academic institutions. However, from a valuation perspective we are mostly concerned with drugs that are in Phase 1 or more advanced and for which better data is available.
A Phase I clinical trial is often less than one year but can take 12-24 months and involves 20-100 individuals. A Phase II trial will typically take 12-24 months and involve a larger number of individuals than in Phase I. A Phase III trial is often in the range of 18-36 months and can involve a significantly larger number of participants.
According to a “Innovation in the pharmaceutical industry: New estimates of R&D costs,” a study published by Joseph A. DiMasi, Henry G. Grabowski, and Ronald W. Hansen in the Journal of Health Economics in 2016, the mean time to advance from start of Phase I to II (including non-trial time) was 19.8 months, the mean time from start of Phase II to III was 30.3 months, and the mean time from start of Phase III to an NDA 30.7 months. The authors further found that the mean approval process time was 16 months, meaning that the overall mean time from start of Phase I to drug approval was 96.8 months or 8.1 years. This study analyzed 106 new drugs from 10 pharmaceutical companies.
Drug Development Success Rates
The development of a new drug is a high-risk endeavor, with the probability of success from initial target identification being less than 1% on average. Once a development project enters the clinical trial stage the likelihood of success improves considerably but is still often in the range of 10% to 20%, depending on product type and therapeutic focus.
Several studies have been performed in the past 30 years on costs and probabilities for clinical trial drug development. One of these is “Trends in risks associated with new drug development: Success rates for investigational drugs,” Nature, 2010, by Joseph A. DiMasi, Lisa R. Feldman, Alain C. Seckler and Alan Wilson. This study analyzed the development histories of 1,738 new drugs from the 50 largest pharmaceutical firms, by clinical phase, therapeutic area, and type of product (small vs. large molecule). The overall average success rate from start of Phase I to NDA or BLA approval was calculate at 19%, with 71% in Phase I, 45% in Phase II, 61% in Phase III, and 93% for NDA/BLA approval. We can also note that the overall success rate for large molecules (biologics including monoclonal antibodies and recombinant proteins) was significantly higher at 32% versus 13% for small molecules.